Blog
Biosimilars 2021 Year in Review
Authors
-
- Name
- Person title
- Principal

-
- Name
- Person title
- Associate

-
- Name
- Person title
- Associate

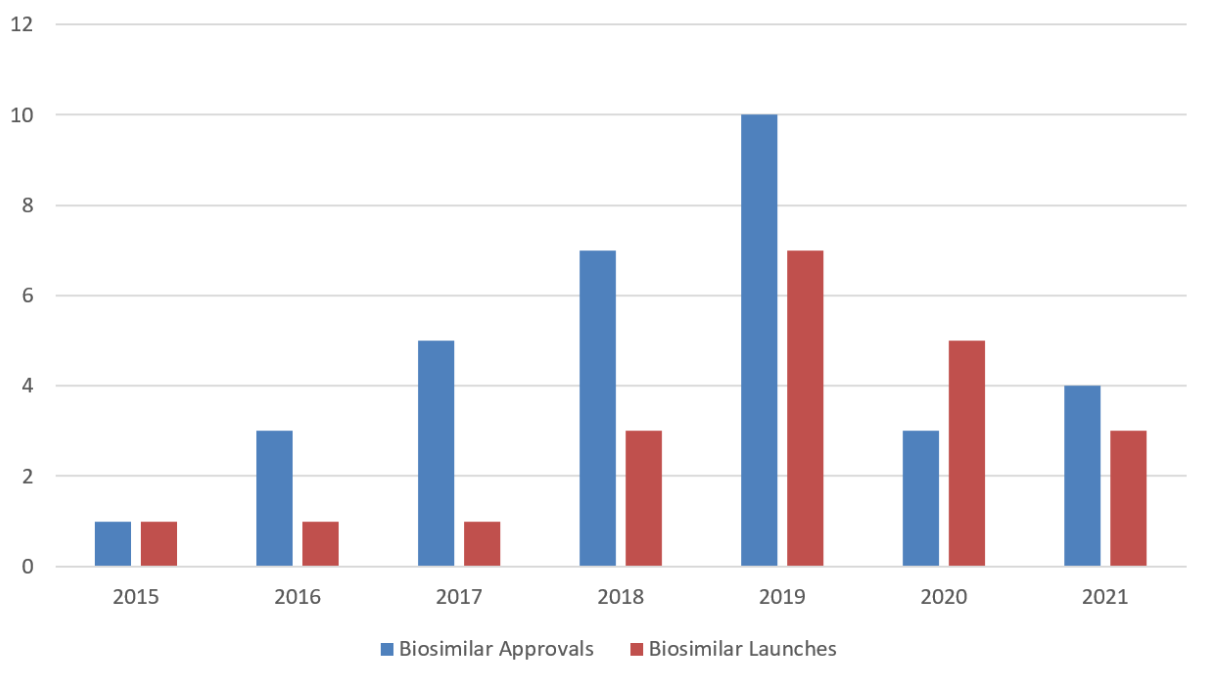
2021 saw several important milestones in the biosimilars space, including the much anticipated first interchangeable designations by FDA and the approval of the first ophthalmology biosimilar. The biosimilar market also exhibited continued growth, with multiple biosimilar developers reporting strong sales of biosimilars through 2021. Yet, given fewer FDA approvals in 2020 and 2021, this year saw the lowest number of commercial launches since 2017. Litigation under the Biologics Price Competition and Innovation Act (BPCIA) in the district courts also decreased.
Biosimilar Approvals and Launches in 2021
Biosimilar Regulatory Updates
Legislation and Executive Orders Relating to Biologics and Biosimilars
BPCIA Litigation
Antitrust and Competition
Post-Grant Challenges at the PTAB
Conclusion
Biosimilar Approvals and Launches in 2021
Similar to last year, 2021 saw relatively few new biosimilar approvals from FDA, with only four newly approved biosimilars: Mylan’s (now Viatris’s) Semglee® and Eli Lilly’s Rezvoglar™, the first and second Lantus® (insulin glargine) biosimilars; Samsung Bioepis’s Byooviz™, the first Lucentis® (ranibizumab) biosimilar; and Coherus’s Yusimry™, a Humira® (adalimumab) biosimilar. The continued slow pace of approvals is potentially the result of the COVID-19 pandemic and related delays in clinical trials and facility inspections by FDA, as well as a re-prioritization and shifting of agency resources towards COVID-19 vaccines and treatments.
The few approvals this year were nevertheless momentous: FDA approved the first interchangeable biosimilar, Semglee® (insulin glargine-yfgn) in July. Then, on October 15, 2021, FDA approved a second interchangeable biosimilar, Boehringer Ingelheim’s (BI) Cyltezo® (adalimumab-adbm) (Cyltezo® was previously approved as a biosimilar in 2017). Several other companies have also announced plans to pursue interchangeable status for biosimilar products. For example, Amgen recently announced that it has Phase 3 studies under way to support interchangeability status for its adalimumab and ustekinumab biosimilars, Amjevita™ and ABP 654, respectively. Pfizer announced plans to pursue interchangeable status for its adalimumab biosimilar Abrilada™ in December 2021. Alvotech has also announced results from a randomized study in patients that demonstrate bioequivalence of repeated switches between administration of Humira® (adalimumab) and Alvotech’s high-concentration proposed biosimilar, AVT02, to administration of Humira® without switching.
With respect to commercial launches, the United States saw fewer new biosimilars entering the market in 2021 than in previous years. Amgen’s Riabni™ (rituximab-arrx) launched in January 2021. Several sources also report that Nyvepria™, Pfizer’s Neulasta® biosimilar, hit the market in late 2020 or early 2021.1 Mylan’s Semglee®, which had previously launched in August 31, 2020,2 became the first interchangeable biosimilar on the United Stated market in November 2021.
But more launches are likely to come as new biosimilars complete clinical testing and FDA review and are approved. The number of development programs enrolled in FDA’s Biosimilar Biological Product Development Program has continued to increase, with 100 biosimilar development programs enrolled as of Q4 of Fiscal Year 2021. Consistent with this, as shown below in Table 2, numerous companies have publicly announced the submission and/or acceptance of new biosimilar BLAs. Companies are also continuing to invest in clinical trials of biosimilars of new compounds, such as Amgen, which is running Phase 3 clinical trials of ABP 938 (a biosimilar to Eylea® (aflibercept)), ABP 654 (a biosimilar to Stelara® (ustekinumab)), and ABP 959 (a biosimilar to Soliris® (eculizumab)).
Figure 1 below provides an overview of biosimilar approvals by FDA and product launches in the United States from 2015 to 2021.
Figure 1. Biosimilar Approvals and Launches by Year
The tables below provide an overview of publicly available information regarding approved and select pending BLAs.
Table 1 summarizes information related to the biosimilars approved as of 2021. FDA has approved 33 biosimilars corresponding to eleven different reference products.
Table 1. Biosimilars Approved as of 2021
| Biosimilar Drug | Reference Product | Biosimilar Code Name | FDA Approval Date | Time from BLA Acceptance to Approval | Commercial Launch Date | Reported Discount at Launch |
| Yusimry™ / CHS-1420 (Coherus) |
Humira® (AbbVie) | adalimumab-aqvh | December 17, 2021 | 12 months | No earlier than 2023 per settlement | |
| Rezvoglar™ (Eli Lilly) |
Lantus® (Sanofi) |
insulin glargine-aglr | December 17, 2021 | 12 months | ||
| Byooviz™ (Samsung Bioepis / Biogen) |
Lucentis® (Roche / Genentech) |
ranibizumab-nuna | September 17, 2021 | 12 months | No earlier than June 2022 | |
| Semglee® (Mylan (Viatris) / Biocon) | Lantus® (Sanofi) |
insulin glargine-yfgn | July 28, 2021 | 31 months | August 31, 2020
Interchangeable: November 2021 |
Unbranded: 65% off WAC of Lantus®
Branded Semglee®: 5% off WAC of Lantus®, with high rebates |
| Riabni™ (Amgen) |
Rituxan® (Roche/ Genentech) |
rituximab-arrx | December 17, 2020 | 12 months | January 12, 2021 | 16.7% off ASP and 23.7% off WAC of Rituxan®; and 15.2% off WAC of Truxima® |
| Hulio® (Mylan) | Humira® (AbbVie) | adalimumab-fkjp | July 6, 2020 | 12 months | No earlier than 2023 per settlement | |
| Nyvepria™ (Pfizer) | Neulasta® (Amgen) | pegfilgrastim-apgf | June 10, 2020 | 12 months | Reported late 2020 / early 2021 | 37% off WAC of Neulasta® |
| Avsola® (Amgen) | Remicade® (Johnson & Johnson) | infliximab-axxq | December 6, 2019 | 12 months | July 2020 | 57% off WAC of Remicade® |
| Abrilada™ (Pfizer) | Humira® (AbbVie) | adalimumab-afzb | November 15, 2019 | 12 months | No earlier than 2023 per settlement | |
| Ziextenzo® (Sandoz) | Neulasta® (Amgen) |
pegfilgrastim-bmez | November 4, 2019 | 50 months (first submission Nov. 2015; resubmitted Feb. 2019) | November 15, 2019 | 37% off WAC of Neulasta® |
| Hadlima® (Samsung Bioepis) |
Humira® (AbbVie) | adalimumab-bwwd | July 23, 2019 | 12 months | No earlier than 2023 per settlement | |
| Ruxience® (Pfizer) |
Rituxan® (Roche / Genentech) |
rituximab-pvvr | July 23, 2019 | 12 months | January 23, 2020 | 24% off WAC of Rituxan® |
| Kanjinti® (Amgen/ Allergan) | Herceptin® (Roche / Genentech) | trastuzumab-anns | June 13, 2019 | 22.5 months
(first submission July 2017; resubmitted Dec. 2018) |
July 18, 2019 | 13% off ASP and 15% off WAC of Herceptin® |
| Zirabev® (Pfizer) |
Avastin® (Roche / Genentech) | bevacizumab-bvzr | June 27, 2019 | 12 months | December 31, 2019 | 23% off WAC of Avastin® |
| Eticovo™ (Samsung Bioepis) |
Enbrel® (Amgen) | etanercept-ykro | April 25, 2019 | 23 months
(first submission May 2017; resubmitted Oct. 2018) |
||
| Trazimera® (Pfizer) |
Herceptin® (Roche / Genentech) | trastuzumab-qyyp | March 11, 2019 | 20.5 months
(first submission June 22, 2017; resubmitted Sept. 28, 2018) |
February 15, 2020 | 22% off WAC of Herceptin® |
| Ontruzant® (Samsung Bioepis/ Merck) | Herceptin® (Roche / Genentech) | trastuzumab-dttb | January 18, 2019 | 15 months | April 15, 2020 | 15% off Herceptin® |
| Herzuma® (Celltrion/Teva) | Herceptin® (Roche / Genentech) | trastuzumab-pkrb | December 14, 2018 | 18.5 months (first submission May 2017; resubmitted June 2018) | March 16, 2020 | 10% off WAC of Herceptin® |
| Truxima® (Celltrion/Teva) | Rituxan® (Roche/ Genentech) |
rituximab-abbs | November 28, 2018 | 19 months
(first submission April 2017; resubmitted May 2018) |
November 11, 2019 | 10% off WAC of Rituxan® |
| Udenyca® (Coherus) | Neulasta® (Amgen) | pegfilgrastim-cbqv | November 2, 2018 | 27 months
(first submission Aug. 2016; resubmitted May 2018) |
January 3, 2019 | 33% off WAC of Neulasta® |
| Hyrimoz® (Sandoz) | Humira® (AbbVie) | adalimumab-adaz | October 30, 2018 | 12 months | No earlier than 2023 per settlement | |
| Nivestym® (Pfizer/Hospira) | Neupogen® (Amgen) | filgrastim-aafi | July 20, 2018 | 10 months | October 1, 2018 | 30.3% off WAC of Neupogen®; 20.3% off WAC of Zarxio®; and 14.1% off WAC of Granix® |
| Fulphila® (Mylan/Biocon) | Neulasta® (Amgen) | pegfilgrastim-jmdb | June 4, 2018 | 18 months (first submission Feb. 2017; resubmitted Dec. 2017) | July 26, 2018 | 33% off WAC of Neulasta® |
| Retacrit® (Pfizer/Hospira) | Epogen®/ Procrit® (Amgen/J&J) | epoetin alfa-epbx | May 15, 2018 | 41 months
(first submission Jan. 2015; resubmitted Dec. 2016) |
November 12, 2018 | 33.5% off WAC of Epogen®; 57.1% off WAC of Procrit® |
| Ixifi® (Pfizer) | Remicade® (Johnson & Johnson) | infliximab-qbtx | December 13, 2017 | 10 months | No U.S. launch intended | |
| Ogivri® (Mylan) | Herceptin® (Roche/ Genentech) |
trastuzumab-dkst | December 1, 2017 | 13 months | December 2, 2019 | 15% off WAC of Herceptin® |
| Mvasi® (Amgen/ Allergan) |
Avastin® (Roche / Genentech) | bevacizumab-awwb | September 14, 2017 | 10 months | July 18, 2019 | 12% off ASP and 15% off WAC of Avastin® |
| Cyltezo® (Boehringer Ingelheim) | Humira® (AbbVie) | adalimumab-adbm | August 25, 2017
Interchangeable: October 15, 2021 |
10 months | No earlier than 2023 per settlement | |
| Renflexis® (Samsung Bioepis/Merck) | Remicade® (Johnson & Johnson) | infliximab-abda | April 21, 2017 | 13 months | July 2017 | 35% off WAC of Remicade® |
| Amjevita™ (Amgen) | Humira® (AbbVie) | adalimumab-atto | September 23, 2016 | 10 months | No earlier than 2023 per settlement | |
| Erelzi® (Sandoz) | Enbrel® (Amgen) | etanercept-szzs | August 30, 2016 | 13 months | ||
| Inflectra® (Pfizer/ Celltrion) |
Remicade® (Johnson & Johnson) | infliximab-dyyb | April 5, 2016 | 20 months
(first submission Aug. 2014; resubmitted Oct. 2015) |
November 2016 | 15% off WAC of Remicade® |
| Zarxio® (Sandoz) | Neupogen® (Amgen) | filgrastim-sndz | March 6, 2015 | 10 months | September 2015 | 15% off WAC of Neupogen® |
Table 2 below shows selected pending biosimilar BLAs for which information is publicly available.
Table 2. Select Pending Biosimilar BLAs as of December 2021
| Proposed Biosimilar | Reference Product | Nonproprietary Name | FDA Status |
| EGI014
(Sandoz / EirGenix) |
Herceptin® (Roche / Genentech) | trastuzumab | BLA Submitted: December 2021 |
| TX-01 (Tanvex) | Neupogen® (Amgen) | filgrastim | BLA Resubmitted: November 22, 2020 |
| TX-05 (Tanvex) | Herceptin® (Roche / Genentech) | trastuzumab | BLA Accepted: October 4, 2021 |
| FYB201/BQ201/CHS-201 (Formycon AG / Bioeq AG / Coherus) | Lucentis® (Roche / Genentech) | ranibizumab | BLA Accepted: October 1, 2021 |
| Alymsys® (Amneal / mAbxience) |
Avastin® (Roche / Genentech) | bevacizumab | BLA Accepted: June 17, 2021 |
| Pegfilgrastim biosimilar (Lupin) | Neulasta® (Amgen) | pegfilgrastim | BLA Accepted: June 2, 2021 |
| BAT1706 (Bio-Thera) | Avastin® (Roche / Genentech) | bevacizumab | BLA Accepted: January 28, 2021 |
| AVT02 (Alvotech) | Humira® (AbbVie) | adalimumab | BLA Accepted: November 2020; delayed due to COVID-19 pandemic |
| MYL-1402O (Mylan / Biocon) | Avastin® (Roche / Genentech) | bevacizumab | BLA Accepted: March 2020
FDA Goal Date: December 27, 2020, but delayed due to COVID-19 pandemic |
| SB8 (Samsung Bioepis) | Avastin® (Roche / Genentech) | bevacizumab | BLA Submitted: September 2019
BLA Accepted: November 2019 |
| Stimufend® (Fresenius Kabi) | Neulasta® (Amgen) | pegfilgrastim | BLA Accepted: May 2020 |
In addition, biosimilar uptake in the United States has continued to trend upwards, with several companies announcing strong biosimilar sales in 2021. It has been reported that filgrastim, trastuzumab, bevacizumab, and rituximab biosimilars have captured well over 50% of their respective markets. Even infliximab biosimilars, which initially had minimal market penetration, reportedly garnered >25% of the infliximab market by the end of Q2 2021.
Back to top ↑II. Biosimilar Regulatory Updates
In 2021, regulatory agencies focused on three interrelated issues: (1) decreasing anti-competitive behavior in the biosimilars space, (2) lowering biologic drug prices, and (3) providing further guidance to clarify the regulatory pathway for biosimilars.
A. Targeting Anti-Competitive Behavior
In February 2020, FDA and the Federal Trade Commission (FTC) issued a joint statement stating that “anti-competitive practices, such as making false or misleading statements comparing biological reference products and biosimilars, may be slowing progress and hampering uptake of these important therapies.” The agencies committed to “take appropriate steps to address companies making false or misleading communications about biologics, including biosimilars and interchangeable products, which will help deter anti-competitive behavior in the biologics market and can help lead to the use of all available biological products.” Citing this joint statement in a July 2021 press release, FDA announced that it had issued an untitled letter to Amgen citing issues with a banner advertisement of its biological product, Neulasta® (pegfilgrastim). In particular, FDA flagged as false or misleading Amgen’s claims in the advertisement that “In a Real-World Study with nearly 11,000 patients Pegfilgrastim PFS resulted in a significantly higher risk of FN [(febrile neutropenia)] vs Onpro®.” According to FDA, Amgen’s violations were “concerning from a public health perspective” because the claims in the advertisement “could cause healthcare providers to conclude that Neulasta delivered via the Onpro on-body injector (OBI) is more effective than Neulasta delivered via prefilled syringe (PFS) or that it is more effective than FDA-licensed biosimilar pegfilgrastim products, which are only delivered via PFS.”
FDA also continued to consider alleged anti-competitive behavior raised in a December 2020 citizen petition filed by BI. BI had asked FDA to change its interpretation of the term “strength” as used in the BPCIA for parenteral solutions to mean “total drug content” without regard to concentration. According to BI’s petition, a change in interpretation was necessary to: “(1) ensure [FDA’s] interpretation is consistent with the clear meaning of the [BPCIA]; (2) prevent abusive ‘evergreening’ tactics from stifling competition of affordable biosimilar and interchangeable biological products; and (3) maintain fair and consistent treatment of all similarly situated parenteral biological products.” BI argued that FDA’s current interpretation “encourages, or at least permits, brand sponsors to use minor concentration changes as an anti-competitive tactic.” FDA granted BI a “listening meeting” on March 17, 2021 in which BI presented its position and answered questions from FDA. On May 28, 2021, FDA notified BI via an interim response that the issues raised in BI’s petition had not yet been resolved “because it raises complex issues requiring extensive review and analysis by Agency officials.”
B. Tackling Biologic Drug Pricing
On September 9, 2021, the U.S. Department of Health & Human Services (HHS) released its Comprehensive Plan for Addressing High Drug Prices. The Plan is replete with suggestions that involve or impact biosimilars. For example, the Plan calls for “[s]upport[ing] market changes that strengthen supply chains, promote biosimilars and generics, and increase transparency.” It also describes various “bold” legislative policies Congress could pursue, such as “[l]egislation to speed the entry of biosimilar and generic drugs, including shortening the period of exclusivity, and policies in Medicare Part B to increase the prescribing of biosimilars by clinicians.” Finally, the Plan summarizes various administrative tools HHS can use to promote competition and reduce drug prices, including “[t]esting models providing additional cost-sharing support to Medicare Part D Low-Income Subsidy Beneficiaries for using biosimilars and generics” and continuing to implement FDA’s Biosimilars Action Plan released in 2018.
On September 10, 2021, Acting FDA Commissioner Janet Woodcock, MD, wrote to Acting Director of the USPTO Andrew Hirshfeld, inviting USPTO to collaborate with FDA in efforts to “advance competition and access in the marketplace” through biosimilars and generics. The letter discusses the BPCIA and the Hatch-Waxman Act frameworks (for small molecule drugs) and sets forth several topics for USPTO’s consideration and further discussion: engagement between FDA and USPTO to increase efficiencies, possible misuse of the patent system (e.g., evergreening or product hopping), adequate time and resources for USPTO Examiners to “ensure the right balance of rewarding innovation and facilitating competition,” and whether the Patent Trial and Appeal Board (PTAB) is addressing patents listed in the Orange and Purple Books.
C. New FDA Guidance and Information
In September 2021, FDA issued two Guidances for Industry: (1) a second revision of a Final Guidance titled, “Questions and Answers on Biosimilar Development and the BPCI Act” and (2) a third revised Draft Guidance titled, “New and Revised Draft Q&As on Biosimilar Development and the BPCI Act.” In the guidance documents, FDA provides “answers to common questions from prospective applicants and other interested parties regarding the Biologics Price Competition and Innovation Act of 2009 (BPCI Act).” The guidance documents are “intended to inform prospective applicants and facilitate the development of proposed biosimilars and interchangeable biosimilars, as well as describe FDA’s interpretation of certain statutory requirements added by the BPCI Act.” FDA revised and withdrew Q&As and provided final answers to the following five questions:
- Q.I.16. How can a proposed biosimilar product applicant fulfill the requirement for pediatric assessments or investigations under [Pediatric Research Equity Act] PREA?
- Q.I.20. What is the nature and type of information that a sponsor should provide to support a post-approval manufacturing change for a licensed biosimilar product?
- Q.I.21. May a sponsor seek approval, in a 351(k) application or a supplement to an approved 351(k) BLA, of a route of administration, a dosage form, or a strength that is different from that of the reference product?
- Q.I.22. May a sponsor seek approval, in a 351(k) application or a supplement to an approved 351(k) BLA, for a condition of use that has not previously been approved for the reference product?
- Q.I.24. May an applicant submit data and information to support approval of a proposed biosimilar or interchangeable product for an indication for which the reference product has unexpired orphan exclusivity?
With respect to Q.I.16, FDA provided guidance regarding the interplay of the PREA and the BPCIA. With respect to Q.I.20, FDA advised that data must be submitted to demonstrate “comparability of the biosimilar product before and after the manufacturing change,” and provided details on the type of data expected. The final answers to Q.I.21 and Q.I.22 are “no.” For Q.I.24, FDA advised that the applicant should submit data to support approval, but FDA will not approve the proposed product for the protected indications.
FDA also began implementing the Biological Product Patent Transparency section of the Consolidated Appropriations Act of 2021 enacted December 27, 2020. As of June 25, 2021 (180 days after enactment), FDA was obligated to publish certain biologic and biosimilar information in a searchable, electronic format. FDA was also obligated to begin publishing patent lists provided by reference product sponsors during the patent dance under § 351(l)(3)(A) of the BPCIA. The Purple Book “FAQs” section has been updated to reflect these new obligations.
FDA further published new fact sheets to provide additional educational materials on biosimilar and interchangeable products and the biosimilar regulatory review and approval process.
III. Legislation and Executive Orders Relating to Biologics and Biosimilars
Federal and state legislators this year have continued to focus on curbing costs of prescription drugs and biologics and invigorating competition in the pharmaceutical space, with potential implications for biosimilars.
A. President Biden’s Executive Order on Promoting Competition in the American Economy
On July 9, 2021, President Biden issued Executive Order 14036 on “Promoting Competition in the American Economy.” The introduction states that “Americans are paying too much for prescription drugs and healthcare services” and points out that “too often, patent and other laws have been misused to inhibit or delay—for years and even decades—competition from generic drugs and biosimilars, denying Americans access to lower-cost drugs.” Among other initiatives, the Order sets out directives to FTC to promulgate rules to prevent “unfair anticompetitive conduct or agreements in the prescription drug industries, such as agreements to delay the market entry of generic drugs or biosimilars.” The Order also directs FDA to address many issues impacting biosimilars, including: (1) “improving and clarifying the standards for interchangeability of biological products”; (2) “supporting biosimilar product adoption by providing effective educational materials and communications to improve understanding of biosimilar and interchangeable products among healthcare providers, patients, and caregivers”; (3) facilitating “development and approval of biosimilar and interchangeable products”; and (4) working with FTC to identify and address “efforts to impede generic drug and biosimilar competition.”
B. Federal Legislation
On April 23, 2021, President Biden signed into law the Advancing Education on Biosimilars Act, which calls for the government to provide educational materials to health care providers, patients, and the general public to increase awareness, knowledge, and confidence in the safety and efficacy of approved biosimilars.
A slew of additional federal legislation related to biosimilars is in the pipeline.
For example, on April 2021, Representatives Paul D. Tonko (D-NY) and Bob Gibbs (R-OH) introduced the Star Rating for Biosimilars Act (H.R. 2855), which would direct HHS to evaluate Medicare plans based on whether cost-saving biosimilars are available to enrollees and to evaluate Medicare Advantage and Part D plans on how they provide access to biosimilars. That same month, Representatives Kurt Schrader (D-OR) and Adam Kinzinger (R-IL) introduced the Bolstering Innovative Options to Save Immediately on Medicines (BIOSIM) Act (H.R. 2815). The Act aims to reduce prices of biologic drugs by providing a temporary increase in reimbursement to the average sales price (ASP) plus 8% (from ASP + 6%, previously) for providers when they use a biosimilar that is lower in price than the reference product.
In July 2021, the U.S. Senate Judiciary Committee passed a legislative package of four bills similarly targeting drug pricing and specific anti-competitive tactics allegedly employed by certain pharmaceutical companies to weaken competition and drive up drug prices. One bill, the Stop Significant and Time-wasting Abuse Limiting Legitimate Innovation of New Generics (Stop STALLING) Act (S. 1425), takes aim at so-called “sham petitions” by making it an unfair method of competition to submit an objectively baseless petition to FDA in an attempt to interfere with a competitor’s application for market approval of a drug. The proposed law would also give FTC enhanced authority to take action against filers of such sham petitions to recover a civil penalty, such as a fine of up to $50,000 for each day the baseless petition was under review, and seek other appropriate relief. Another bill, the Affordable Prescriptions for Patients Act (S. 1435), would amend the FTC Act to prohibit drug manufacturers from engaging in so-called “hard switch” or “soft switch” product hopping. The Preserve Access to Affordable Generics and Biosimilars Act (S. 1428) would amend the FTC Act to make anticompetitive “pay-for-delay” (also known as “reverse-payment”) settlement agreements that prevent or delay the introduction of generic drugs or biosimilars presumptively illegal. Finally, the Prescription Pricing for the People Act (S. 1388) would require the FTC to study the role of intermediaries such as pharmacy benefit managers in the pharmaceutical supply chain and provide Congress with “appropriate” policy recommendations to increase transparency in the supply chain and prevent anticompetitive practices that may impact the cost of prescription drugs.
In September 2021, the House Judiciary Committee advanced a similar set of competition- and pricing-related bills to the full House, including the Stop Stalling Access to Affordable Medications Act (H.R. 2883), the Affordable Prescriptions for Patients Through Promoting Competition Act (H.R. 2873), and the Preserve Access to Affordable Generics and Biosimilars Act (H.R. 2891), companion bills to S. 1425, S.1435, and S. 1428, respectively. The House also advanced the Affordable Prescriptions for Patients Through Improvements to Patent Litigation Act (H.R. 2884), which would limit in certain instances the number of patents that a reference product sponsor could assert against a biosimilar applicant in a lawsuit following the BPCIA’s “patent dance.”
C. State Legislation
Starting in 2013, state legislators nationwide have enacted laws regulating biosimilar substitution. This April, Oklahoma Governor Kevin Stitt signed into law SB 4, making Oklahoma the final state to enact a biosimilar substitution law. Like similar legislation in other states, SB4 allows for interchangeable biosimilars to be substituted at the pharmacy once approved by FDA and requires the pharmacy to inform patients and physicians of a substitution.
Many states have also focused on decreasing prescription drug costs and increasing competition through biosimilars. For example, in February 2021, Minnesota lawmakers introduced a new bill, SF 990, designed to expand Minnesota consumers’ access to biosimilars. The proposed legislation would require health plans and pharmacy benefit managers to cover all versions of biological drugs, including biosimilars.
Other states have taken aim at anticompetitive settlements allegedly delaying access to biosimilars. California Assembly Bill 824 (AB 824) went into effect on January 1, 2020. AB 824 was a first-of-its-kind state law that regulated anti-competitive “pay-for-delay” agreements, when brand name pharmaceutical companies or reference product sponsors (RPS) pay generic drug or biosimilar product manufacturers to slow down or stop lower-cost medications from entering the market. The most notable features of California’s law are its presumption that such settlements are anticompetitive and unlawful if the generic or biosimilar received “anything of value,” and its provision for civil penalties of at least $20 million. The Association for Accessible Medicines (AAM) immediately challenged the constitutionality of AB 824 in the Eastern District of California. AAM’s initial lawsuit concluded after the Ninth Circuit found that AAM lacked standing to bring suit on its members’ behalf due to its failure to show that there was a “substantial risk” that its members faced harm as a result of the challenged law. Ass’n for Accessible Medicines v. Becerra, 822 F. App’x 532 (9th Cir. 2020). After bringing a follow-on lawsuit again challenging AB 824 in August 2020, AAM filed a new motion for a preliminary injunction, which was granted by Judge Troy L. Nunley in an order dated December 9, 2021. Ass’n for Accessible Medicines v. Bonta, No. 2:20-cv-01708, 2021 WL 5853431 (E.D. Cal. Dec. 9, 2021). The court found a likelihood of success based on AAM’s constitutional challenge under the dormant Commerce Clause (regulating activity “wholly outside” California). The court enjoined enforcement of AB 824.
Despite the challenges faced by AB 824, several states have introduced bills that are directed to curbing anti-competitive pay-for-delay agreements entered into by pharmaceutical companies. These bills, which are currently pending in their respective state legislatures, include Oregon SB 764, Connecticut SB 269, Illinois HB 3729, Minnesota SF 184, and New York S398. Many have similarities to the California law, and AAM has already come out in opposition to some, such as Oregon’s SB 764. The fate of these bills remains uncertain.
IV. BPCIA Litigation
Below, we briefly summarize overall statistics regarding BPCIA district court litigation to-date, and in the subsequent sections, we review (a) ongoing BPCIA district court cases, (b) BPCIA district court cases that settled or resolved in 2021, and (c) non-BPCIA district court and Federal Circuit decisions that bear on biologics and biosimilars. Note that the Federal Circuit did not decide any BPCIA appeals in 2021, and there were no BPCIA appeals pending before the Federal Circuit at the end of 2021.
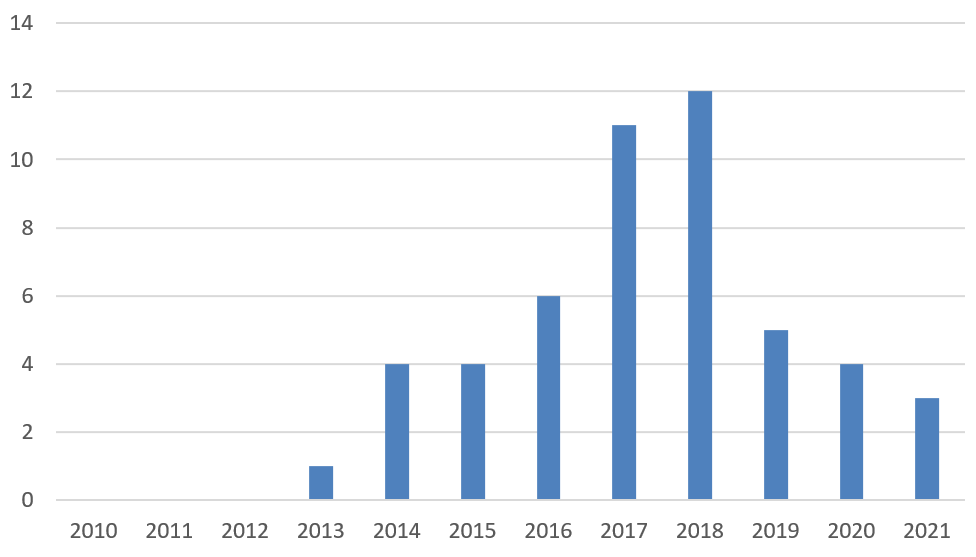
Since the BPCIA’s enactment in 2010, 50 BPCIA cases have been filed in district courts. (See Figure 2.) Many of these cases involve the same parties and the same biosimilar products, so do not reflect unique disputes. Amgen (with 16 cases) and Genentech (with 15 cases) are the most active plaintiffs and together account for the plaintiff side in more than half of all BPCIA litigation. Amgen is also the most common BPCIA defendant (with 8 cases), while Celltrion (defendant in 7 cases) and Sandoz (defendant in 6 cases) are not far behind. AbbVie has also been active in BPCIA litigation with 6 cases (plaintiff in 5 cases and declaratory judgment defendant in 1 case).
Figure 2. BPCIA Cases Filed by Year since BPCIA Enactment
BPCIA litigation has involved biosimilars of nine different reference products: Remicade®, Neulasta®, Neupogen®, Avastin®, Herceptin®, Rituxan®, Humira®, Enbrel®, and Epogen®. Unsurprisingly, these mirror the nine reference products for which there were FDA-approved biosimilars prior to 2021 (see Section I, above). In the second half of 2021, FDA approved biosimilars referencing Lucentis® and Lantus® (see Table 1, above). There has not yet been BPCIA litigation involving biosimilars of these two new reference products.
In 2019, BPCIA district court filings began to decrease, and this trend continued in 2020 and 2021. (See Figure 2.) Whereas 2018 had a record 12 BPCIA district court filings, 2019 had five, 2020 had four, and 2021 had only three. We now turn to these three new cases and other recent litigation activity.
A. Ongoing BPCIA District Court Litigation
All three BPCIA cases filed in 2021 (identified in Table 3) involved the same biosimilar product, AVT02, referencing Humira® (adalimumab).
Table 3. BPCIA Cases Filed in 2021
| Case Name | Court | Filing Date | Biosimilar at Issue | Reference Product at Issue | Type of Complaint | # Asserted Patents |
| AbbVie v. Alvotech hf. (1:21-cv-02258) |
N.D. Ill. | 4/27/2021 | AVT02 | Humira® (adalimumab) |
Patent infringement | 4 |
| Alvotech USA et al. v. AbbVie (1:21-cv-05645) |
E.D. Va. / N.D. Ill. | 5/11/2021 | AVT02 | Humira® (adalimumab) |
Declaratory judgment | 4 |
| AbbVie v. Alvotech hf. (1:21-cv-02899) |
N.D. Ill. | 5/28/2021 | AVT02 | Humira® (adalimumab) |
Patent Infringement | 58 (now 61) |
Both patent infringement cases (-2258, -2899) remain ongoing. The declaratory judgment action (-5645) was dismissed without prejudice in November 2021.
In addition, two cases filed prior to 2021 remain ongoing:
- Amgen v. Hospira (20-cv-00201 D. Del.); Neulasta®/Nyvepria™
- Genentech v. Samsung Bioepis (20-cv-00859 D. Del.); Avastin®/SB8
We discuss each ongoing BPCIA district court case briefly below.
AbbVie v. Alvotech hf. (21-cv-02258 N.D. Ill.; 21-cv-02899 N.D. Ill.); Alvotech USA et al. v. AbbVie (21-cv-05645 N.D. Ill.)
This series of cases involves Alvotech’s AVT02, a proposed biosimilar of AbbVie’s high-concentration (100 mg/mL) Humira® (adalimumab). The -2258 and -2899 cases involve only Alvotech hf., an Icelandic company that houses a biopharmaceutical facility involved in manufacturing AVT02. The -5645 case involved both Alvotech hf. and its subsidiary, Alvotech USA, a Virginia-based company responsible for “legal, governmental policy, and regulatory affairs.” (See, e.g., -5645 Dkt. 1 at ¶¶ 20–21.) Alvotech USA “is the applicant for FDA biologics license application (‘BLA’) No. 761205 under 42 U.S.C. § 262(k)” for AVT02. (Id. at ¶ 22.) Alvotech USA and AbbVie engaged in the patent dance from November 2020 through April 2021, resulting in four patents identified for the first wave litigation. (Id. at ¶¶ 29–37.)
On April 27, 2021, AbbVie sued the parent company, Alvotech hf., in the Northern District of Illinois alleging infringement of the four patents that resulted from the patent dance. (-2258 Dkt. 1 at ¶¶ 61–62.) This was the first BPCIA litigation filed in the Northern District of Illinois, but the fourth BPCIA litigation brought by AbbVie against an adalimumab biosimilar manufacturer, following litigation against Amgen, Sandoz, and BI, which settled in 2017, 2018, and 2019, respectively.
Shortly thereafter, on May 11, 2021, Alvotech USA provided AbbVie with Notice of Commercial Marketing pursuant to 42 U.S.C. § 262(l)(8)(A), and Alvotech USA and Alvotech hf. jointly filed a declaratory judgement action against AbbVie in the Eastern District of Virginia (originally 1:21-cv-00589 E.D. Va.). (-5645 Dkt. 1 at ¶ 41.) In their declaratory judgment complaint, the Alvotech entities alleged that “AbbVie Inc. has attempted to disadvantage Alvotech USA by filing suit against Alvotech hf. in the Northern District of Illinois, and not suing Alvotech USA, the subsection (k) applicant.” (Id. at ¶ 52.) The Alvotech entities also included allegations that AbbVie’s patents were unenforceable due to inequitable conduct and unclean hands. Among other allegations, the Alvotech entities asserted that “AbbVie’s ‘minefield of IP’ is a collection of highly-dubious patents, at least 60 of which have been used to threaten every would-be competitor, including Alvotech” and ultimately AbbVie’s unclean hands with regard to its patent portfolio renders each patent-in-suit unenforceable. (Id. at ¶ 141.)
On May 28, 2021, AbbVie filed another suit (the -2899 case) against Alvotech hf. in the Northern District of Illinois. In its complaint, AbbVie stated that Alvotech USA’s Notice of Commercial Marketing triggered AbbVie’s ability to file suit on all patents identified in AbbVie’s (3)(A) list during the patent dance. (-2899 Dkt. 1 at ¶ 17.) AbbVie initially asserted 58 additional patents, but subsequently amended its complaint twice to add newly issued patents. (-2899 Dkt. 77, 130.)
On June 2, 2021, Alvotech hf. filed a motion to dismiss the -2258 case, arguing, inter alia, that: (1) the court lacked subject matter jurisdiction and the complaint did not state a claim under the BPCIA because AbbVie failed to sue Alvotech USA, the subsection (k) applicant for the AVT02 biosimilar at issue (and instead sued Alvotech USA’s Icelandic parent company, Alvotech hf.); (2) AbbVie’s complaint failed to name a necessary party, Alvotech USA; and (3) AbbVie’s complaint should be dismissed for lack of personal jurisdiction over Alvotech hf. (-2258 Dkt. 27.) In opposition, AbbVie argued that (1) Alvotech hf. was a proper defendant because it was the “submitter” of the AVT02 BLA; (2) Alvotech USA was not a necessary party to the litigation; and (3) the court had personal jurisdiction over Alvotech hf. (-2258 Dkt. 31.) On August 23, 2021, the court denied Alvotech hf.’s motion to dismiss, finding that (1) “Abbvie’s complaint adequately alleges that Alvotech hf. is a ‘submit[ter]’ of the aBLA within the meaning of 35 U.S.C. § 271(e)(2)(C)”; (2) “Alvotech hf. [] failed to meet its burden of establishing that this case must be dismissed for failing to join Alvotech USA”; and (3) the court has specific jurisdiction “because the aBLA submission indicates Alvotech hf.’s intent to market and distribute its biosimilar drug in Illinois.” (-2258 Dkt. 51 at 20, 24, 28.)
On the same day that Alvotech hf. filed its motion to dismiss the -2258 case, AbbVie filed a motion to dismiss, or in the alternative, to transfer, the case pending in the Eastern District of Virginia (-5645 case). AbbVie argued that: (1) the action should be dismissed or transferred under the first-to-file rule; (2) alternatively, the action should be dismissed for lack of personal jurisdiction; and (3) alternatively, the case should be transferred to Illinois. (-5645 Dkt. 43.) In opposition, the Alvotech entities argued that: (1) dismissal or transfer under the first-filed rule was improper because this action was the first and only action with all the relevant parties; (2) the court had specific personal jurisdiction over AbbVie Inc. and AbbVie Biotechnology Ltd.; and (3) AbbVie’s alternative request to transfer the case to Illinois should be denied. (-5645 Dkt. 44.) On October 22, 2021, the court granted AbbVie’s motion to transfer to the Northern District of Illinois and found the motion to dismiss moot. (-5645 Dkt. 51.) On November 1, 2021, the Alvotech entities filed a notice of voluntary dismissal of the -5645 case. (Dkt. 55.) The court dismissed the case without prejudice on November 5, 2021. (Dkt. 56.)
This left the two cases pending in the Northern District of Illinois against only Alvotech hf.: the -2258 case and the -2899 case. In September 2021, the court ordered a trial on ten patents (three from the -2258 case and seven from the -2899 case) starting on August 1, 2022. (-2258 Dkt. 63; see also Dkt. 54.) The court stated it plans to issue a trial decision by the end of October of 2022, and, “[i]n light of that, [Alvotech] agreed not to launch AVT02 in the United States prior to the issuance of the Court’s decision.” (-2258 Dkt. 63 at 4.) The ten patents span technologies including autoinjector devices, methods of treatment, antibody purification, and pharmaceutical formulations. The parties have recently finished claim construction briefing and are currently in the midst of fact discovery.
Additional motions remain pending. AbbVie has an outstanding motion, filed October 5, 2021, to dismiss and strike Alvotech hf.’s counterclaims and affirmative defenses of inequitable conduct, unclean hands, and patent misuse, arguing that Alvotech hf. failed to adequately plead each of these counterclaims and affirmative defenses. (-2258 Dkt. 70.) Alvotech hf. has an outstanding motion to dismiss the -2899 case pursuant to Federal Rules of Civil Procedure 12(b)(1)–(2) and (6)–(7). (See -2289 Dkt. 29, 91.) The court has not yet issued its claim construction order.
The parties are also embroiled in other litigation related to adalimumab. AbbVie filed a trade secret complaint against Alvotech hf. in the Northern District of Illinois on March 19, 2021. (1:21-cv-01530 N.D. Ill.) AbbVie alleged that “Alvotech embarked on an unlawful plot to surreptitiously take AbbVie’s confidential and proprietary trade secrets related to the confidential large scale manufacturing process for HUMIRA® in order to develop and manufacture its copycat product [AVT02].” (-1530 Dkt. 1 at ¶ 3.) This district court complaint was dismissed for lack of personal jurisdiction on October 6, 2021. (-1530 Dkt. 38.) AbbVie recently asserted similar allegations in an ITC action, filed December 17, 2021, against Alvotech USA, Alvotech hf. and multiple other entities. (See Certain Adalimumab, Processes for Manufacturing or Relating to Same, and Products Containing Same, Inv. No. 337-TA-3585.)
Amgen v. Hospira (20-cv-00201 D. Del.)
This case involves Hospira’s biosimilar of Amgen’s Neulasta® (pegfilgrastim). Amgen filed a complaint in the District of Delaware against Hospira and Pfizer on February 11, 2020, asserting U.S. Patent No. 8,273,707 (the ’707 patent), which is directed to methods of protein purification requiring a certain combination of salts at certain concentrations. (Dkt. 1 at ¶¶ 8, 54, 61–62.) Amgen previously asserted this patent in BPCIA litigation against two other biosimilar developers, Coherus and Mylan, and both of those cases were resolved in 2019.
In the current case, Hospira and Pfizer moved to dismiss, arguing that there is no literal infringement because (1) the accused process uses salt concentrations below those claimed in the ’707 patent (as properly construed in light of alleged disclaimers in the prosecution history), and (2) prosecution history estoppel precludes Amgen from asserting infringement under the doctrine of equivalents against different concentrations of the claimed salts for the same reasons as in the Coherus case. (Dkt. 19 at 1–4.) In opposition, Amgen argued that the Coherus decision “was based on the identity of the salts employed, not their concentration.” (Dkt. 28 at 6.) Amgen also argued that a finding of non-infringement would be premature at the pleading stage because of outstanding claim construction and factual issues, such as the claim construction of “about 0.1 M.” (Id. at 4.) Hospira and Pfizer, in reply, contested Amgen’s positions. (See Dkt. 32.)
On March 23, 2021, the court heard oral argument on the motion to dismiss and entered an oral order denying the motion “[f]or the reasons stated by the Court during [the] telephonic argument.” As of early January 2022, a transcript of the oral argument was not available on the docket.
In a joint claim construction brief filed on March 15, 2021, the parties disputed the constructions of “five groups of related or identical terms.” (Dkt. 62 at 1.) To streamline claim construction, however, the parties jointly proposed that the court “construe first the claim term ‘between about 0.1 M and about 1.0’ and defer construction of the other disputed terms.” (Dkt. 68 at 1.) The parties agreed that construction of the “concentration range term” is “central to the parties’ disputes.” (Id.) Amgen also acknowledged that “the resolution of the proper construction of the [concentration range term] may be dispositive.” (Dkt. 72 at 1.) On June 16, 2021, the court construed the term “between about 0.1 M and about 1.0” in claims 1 and 10 of the ’707 patent “to have its plain and ordinary meaning (that is, ‘between approximately 0.1 M and approximately 1.0 M’) and is not a functional limitation.” (Dkt. 74 at 2.) In light of this construction, Amgen confirmed it would only seek a finding of infringement under the doctrine of equivalents, not literal infringement. (Dkt. 75 at 1.)
On August 6, 2021, Hospira and Pfizer filed a motion for summary judgment of non-infringement, arguing that there is no infringement under the doctrine of equivalents because the accused manufacturing process uses a “low” salt concentration below the claimed “intermediate” range. (Dkt. 84 at 7–8.) Amgen responded that genuine disputes of material fact precluded summary judgment. (Dkt. 91 at 2.) While fully briefed, the court has not yet ruled on Hospira and Pfizer’s motion for summary judgment of non-infringement.
A jury trial is set for July 11, 2022. (Dkt. 33.)
Genentech v. Samsung Bioepis (20-cv-00859 D. Del.)
This case relates to Samsung Bioepis’s proposed biosimilar of Genentech’s Avastin® (bevacizumab). Genentech filed a complaint against Samsung Bioepis on June 28, 2020, alleging infringement of 14 patents covering methods of manufacturing bevacizumab and methods of treating patients with bevacizumab. (Dkt. 1.) Samsung Bioepis is the third bevacizumab biosimilar developer against which Genentech has initiated BPCIA litigation, following litigation against Pfizer and Amgen, which settled in 2019 and 2020, respectively.
On September 21, 2020, Genentech filed a motion to dismiss and strike Samsung Bioepis’s counterclaims and third affirmative defense, arguing that Samsung Bioepis could not pursue patent invalidity arguments that it did not identify in its patent dance disclosures. (Dkt. 12 at 1.) In Genentech’s filings in support of its motion and in Samsung Bioepis’s opposition, the parties disputed the breadth and import of the court’s decision in Genentech, Inc. v. Amgen Inc., No. 17-cv-01407, 2020 WL 636439, at *4–5 (D. Del. Feb. 11, 2020), which held that Amgen was not limited to its invalidity positions set forth during the patent dance. For example, Genentech argued that a biosimilar should not be able to expand its invalidity theories absent good cause and that failing to present invalidity arguments during the dance can result in “forfeiture.” (Dkt. 12 at 5.) We have been unable to locate a ruling on this motion on the public docket.
On February 10, 2021, the parties filed a sealed stipulation staying the action. (Dkt. 29.) The court ordered the stay on February 11, 2021. (Dkt. 32.) A subsequent filing suggested that the stay was requested “on account of the difficulties Bioepis has faced in obtaining FDA approval.” (Dkt. 38 at 4.)
The parties’ stay carved out a dispute related to the protective order and limited use of Bioepis’s confidential information for a Danish proceeding. (See, e.g., Dkt. 38 at 4.) While the parties have continued to file papers related to this discrete issue, the case has been otherwise stayed.
B. BPCIA Litigation Settled or Resolved in 2021
A number of BPCIA cases settled or otherwise resolved in 2021. As mentioned above, Alvotech USA et al. v. AbbVie (21-cv-05645 N.D. Ill., originally in E.D. Va.) was filed and dismissed in 2021. Additional cases resolved in 2021 include:
- Amgen v. Hospira (18-cv-01064 D. Del.); Neupogen®/Nivestym®
- Amgen v. Hospira (20-cv-00561 D. Del.); Neupogen®/Nivestym®
- Immunex v. Sandoz (16-cv-01118 D.N.J.); Enbrel®/Erelzi®
- Immunex v. Samsung Bioepis (19-cv-11755 D.N.J.); Enbrel®/Eticovo™
- Genentech v. Centus/Fujifilm (20-cv-00361 E.D. Tex.); Avastin®/FKB238
We discuss each of these resolved cases briefly below.
Amgen v. Hospira (18-cv-01064 D. Del.)
This case involved Nivestym®, Hospira and Pfizer’s biosimilar of Amgen’s Neupogen® (filgrastim). A trial had been scheduled for May 17, 2021, but the parties requested a continuance in light of the ongoing COVID-19 pandemic. (Dkt. 250.) The court rescheduled the trial for September 20, 2021 (March 16, 2021 Oral Order), but the case was dismissed in advance of that date. On September 1, 2021, the parties filed a stipulation to dismiss all claims and counterclaims in the litigation with prejudice, with each party to bear its own costs, expenses, and attorneys’ fees. (Dkt. 332.) The court entered the order of dismissal on September 8, 2021.
Before the case was dismissed, the parties filed a flurry of motions, including several Daubert and summary judgment motions, a motion by Amgen to strike certain defenses (Dkt. 299), and a motion by Hospira and Pfizer to amend their Answer to add an additional defense (Dkt. 317). For example, Hospira and Pfizer filed motions for summary judgment of invalidity and noninfringement of the asserted claims of U.S. Patent No. 9,643,997 (the “’997 patent”), which relates to methods of purifying proteins expressed in nonmammalian expression systems (e.g., E. coli). (Dkt. 258, 266.) Amgen filed a motion for partial summary judgment of no invalidity of the asserted claims of the ’997 patent. (Dkt. 281.) The court denied Hospira and Pfizer’s motion for summary judgment of noninfringement and Amgen’s motion for partial summary judgment of no invalidity. (Dkt. 296, 307.) With respect to Hospira and Pfizer’s motion for summary judgment of invalidity, the court found that the motion could not be resolved “without first construing the term ‘refold’ as it is used in claim 9 of the asserted patent,” set a hearing for the purpose of construing the term “refold,” and allowed the parties to submit letter briefs on this issue. (Dkt. 312.) This was not resolved prior to the conclusion of the case.
Amgen v. Hospira (20-cv-00561 D. Del.)
This case also involved Nivestym® and was a follow-on to litigation between the same parties discussed above (18-cv-01064 D. Del.). This case involved a newly issued patent—U.S. Patent No. 10,577,392—asserted against the same Neupogen® biosimilar. (Dkt. 1 at ¶ 11.) This case had been stayed pending resolution of the 2018 case. (Dkt. 30.) On September 1, 2021, the parties filed a stipulation to dismiss all claims and counterclaims in the litigation with prejudice, with each party to bear its own costs, expenses, and attorneys’ fees. (Dkt. 32.) The court entered the order of dismissal on September 8, 2021.
Immunex v. Sandoz (16-cv-01118 D.N.J.)
This case involved Erelzi®, Sandoz’s biosimilar of Immunex’s Enbrel® (etanercept). On August 9, 2019, the court held U.S. Patent No. 8,063,182, directed to etanercept, the active ingredient in Enbrel®, and U.S. Patent No. 8,163,522, directed to Enbrel®’s manufacturing process, not invalid. Immunex Corp. v. Sandoz Inc., 395 F. Supp. 3d 366 (D.N.J. 2019). On July 1, 2020, the Federal Circuit affirmed. Immunex Corp. v. Sandoz Inc., 964 F.3d 1049 (Fed. Cir. 2020). On September 29, 2020, the Federal Circuit denied Sandoz’s petition for panel rehearing and rehearing en banc.
(No. 20-1037 at Dkt. 117.) On May 17, 2021, the Supreme Court denied Sandoz’s petition for certiorari. Sandoz Inc. v. Immunex Corp., 141 S. Ct. 2623 (Mem) (2021).
On July 23, 2021, the clerk granted in part and denied in part Immunex’s motion to tax costs against Sandoz pursuant to Federal Rule of Civil Procedure 54(d) and Local Civil Rule 54.1 in light of the fact that it is “undisputed that these Plaintiffs are the prevailing parties.” (Dkt. 761; see also Dkt. 762.) Pursuant to Federal Rule of Civil Procedure 58(b), the clerk granted costs to Immunex in the amount of $109,746.60. (Dkt. 761, 762)
Immunex v. Samsung Bioepis (19-cv-11755 D.N.J.)
This case involved Samsung Bioepis’s biosimilar Eticovo™ referencing Immunex’s Enbrel® (etanercept). The five patents Immunex asserted against Samsung Bioepis in this case (Dkt. 1 at ¶ 6) overlap with those asserted against Sandoz in a case discussed above (16-cv-01118 D.N.J.). The court entered a final judgment in favor of Immunex and an order of permanent injunction on November 3, 2021.
The court had previously entered a consent injunction order in January 2020, prohibiting Samsung Bioepis from making, using, importing, offering to sell, or selling “Bioepis’s Etanercept Product or any other product containing the fusion protein known as etanercept in the United States, except as allowed by [the] 35 U.S.C. § 271(e)(1)” safe harbor. (Dkt. 113 at 1.) The basis for the order was “the Stipulation submitted to the Court on December 23, 2019, D.I. 105” (id.), which is sealed and not publicly available. The case was also administratively stayed consistent with that confidential stipulation. (Dkt. 116.)
On November 2, 2021, the parties submitted a letter requesting that the court enter another confidential stipulation, which is sealed and not publicly available, along with a “Final Judgment and Order of Permanent Injunction.” (Dkt. 126, 132.) On November 3, 2021, the court entered the parties’ confidential stipulation (Dkt. 127) and the Final Judgment and Order of Permanent Injunction, which entered judgment in favor of Immunex on each claim for infringement of U.S. Patent Nos. 8,063,182 (the “’182 patent”) and 8,163,522 (the “’522 patent”); a permanent injunction against Samsung Bioepis making, using, importing, offering to sell, or selling any product containing etanercept, except as allowed by the 35 U.S.C. § 271(e)(1) safe harbor, until the expiration of the ’182 and ’522 patents on April 24, 2029, and requiring Samsung Bioepis to “immediately destroy any remaining Bioepis etanercept product that has been imported into the United States”; and dismissal of Immunex’s claims for infringement of the remaining three asserted patents without prejudice. (Dkt. 128.)
Genentech v. Centus/Fujifilm (20-cv-00361 E.D. Tex.)
This case, involving Centus’s proposed biosimilar, FKB238, referencing Genentech’s Avastin® (bevacizumab), was the first BPCIA litigation filed in the Eastern District of Texas and the first BPCIA litigation involving Centus and its co-defendants Fujifilm Kyowa Kirin Biologics Co., Ltd., Fujifilm Corp., and Kyowa Kirin Co., Ltd.
On April 14, 2021, before the defendants answered the complaint, the parties filed a joint motion to stay all deadlines and a notice of settlement. (Dkt. 16.) The motion stated that “[a]ll matters in controversy between the parties [had] been settled, in principle,” and the parties were in the process of preparing settlement documents. (Id. at 1.) After a short stay, on June 21, 2021, Genentech filed a motion to dismiss with prejudice under Rule 41(a)(1). (Dkt. 23). On July 2, 2021, the court granted the motion to dismiss. (Dkt. 25.)
C. Other District Court Litigation and Federal Circuit Appeals
Although not BPCIA decisions, several Federal Circuit and other district court decisions issued in 2021 may have implications in the biosimilars context. We briefly identify some of these decisions below.
The Federal Circuit has issued numerous high-profile opinions addressing § 112 requirements that could impact biosimilars in the future. For example, Amgen Inc. v. Sanofi, Aventisub LLC, 987 F.3d 1080 (Fed. Cir. 2021) relates to Amgen’s Repatha® (evolocumab), an antibody product for treating high cholesterol. The Federal Circuit held that functionally defined claims covering a genus of antibodies were not enabled because undue experimentation would be required to practice the full scope of the claims. Id. at 1088. The relevant claims defined the claimed antibodies by their function: “binding to a combinations of sites (residues) on the PCSK9 protein, in a range from one residue to all of them; and blocking the PCSK9/LDLR interaction.” Id. at 1083. The court explained that functional limitations “pose high hurdles in fulfilling the enablement requirement for claims with broad functional language.” Id. at 1087. The court also concluded that the functional limitations here were “broad,” “the disclosed examples and guidance [were] narrow,” the invention was “in an unpredictable field of science with respect to satisfying the full scope of the functional limitations,” and “no reasonable jury could conclude under these facts that anything but ‘substantial time and effort’ would be required to reach the full scope of claimed embodiments.” Id. at 1087–88. On June 21, 2021, the court denied Amgen’s petition for panel rehearing and rehearing en banc. Amgen Inc. v. Sanofi, Aventisub LLC, 850 F. App’x 794 (Mem) (Fed. Cir. 2021). Judge Lourie, joined by Judges Prost and Hughes, authored a separate opinion on the denial of the petition for panel rehearing to reject Amgen’s argument that the court created a new test for enablement. Id. Amgen filed a petition for certiorari on November 22, 2021 (No. 21-757).
The Federal Circuit addressed written description in Juno Therapeutics, Inc. v. Kite Pharma, Inc., 10 F.4th 1330 (Fed. Cir. 2021), relating to chimeric antigen receptor (CAR) T-cell therapies. The Federal Circuit reversed a jury award of over $1.2 billion because the jury’s written description verdict was not supported by substantial evidence. Id. at 1332. The claims at issue were genus claims that used functional language (binding function of single-chain variable fragments (scFvs), a part of the CAR that determines what target molecule or antigen the CAR can recognize and bind to). Id. at 1333–34, 1335. The Federal Circuit pointed out that the specification identified only two exemplary scFvs for two different targets, which “[did] not provide information sufficient to establish that a skilled artisan would understand how to identify the species of scFvs capable of binding to the limitless number of targets as the claims require.” Id. at 1337. Further, “[e]ven accepting that scFvs were known and that they were known to bind, the specification provides no means of distinguishing which scFvs will bind to which targets.” Id. at 1338. The Court also found that the specification “[did] not disclose structural features common to the members of the genus to support that the inventors possessed the claimed invention,” for example those structural features that distinguish between scFvs that bind and those at do not. Id. at 1338–39. On October 27, 2021, Juno petitioned for a panel rehearing or rehearing en banc. (No. 20-1758 at Dkt. 81.) Kite responded on December 29, 2021. (No. 20-1758 at Dkt. 100.)
Meanwhile, district courts have grappled with whether specific applications appropriately transitioned (or did not transition) from an NDA to a BLA under the “deemed to be a BLA” provision that went into effect in March 2020.
One such case was decided the very last day of 2020. In Teva Pharmaceuticals USA, Inc. v. United States Food & Drug Administration, 514 F. Supp. 3d 66 (D.D.C. 2020), a case brought under the Administrative Procedure Act (APA), the District Court for the District of Columbia upheld FDA’s interpretation of “protein” under the BPCIA and FDA’s determination that Teva’s glatiramer acetate product, Copaxone®, is not a “biological product.” (As a result, it did not transition in March 2020 from an NDA to a BLA under the “deemed to be a BLA” provision of the BPCIA.) The court found that FDA’s interpretation of the term “protein” to “refer only to molecules that … have a ‘specific, defined sequence’ of amino acids” was reasonable and therefore owed deference under Chevron. Id. at 102, 106. The court also found that “FDA acted reasonably . . . in finding that Copaxone is not a protein” because Copaxone® exhibits “sequence variability” (i.e., its amino acid sequences are “determined during the chemical solution polymerization process”) and thus Copaxone® “did not satisfy the ‘specific, defined sequence’ requirement ….” Id. at 106–07.
In Ipsen Biopharmaceuticals, Inc. v. Becerra, No. 20-cv-2437, 2021 WL 4399531 (D.D.C. Sept. 24, 2021), the District Court for the District of Columbia found that it lacked subject-matter jurisdiction over Ipsen’s complaint, which challenged “FDA’s failure to regulate [] Somatuline Depot as a biological product.” Id. at *3. Ipsen argued that although FDA initially approved Somatuline Depot as a drug, “the substance also meets the recently-amended definition of ‘biological product’” because it is a “‘protein’ within the meaning of the PHSA—i.e., an ‘alpha amino acid polymer with a specific, defined sequence that is greater than 40 amino acids in size.’” Id. at *2. FDA had rejected Ipsen’s arguments, stating that “the proper frame of reference for applying its definition of ‘protein’ is a drug’s ‘active ingredient,’” a “protein” must be “at least 40 amino acids,” and the active ingredient in Ipsen’s product “contains only 8 amino acids.” Id. at *3. Ipsen disagreed—arguing that the frame of reference should be the “finished dosage form” that is marketed and sold—and contended that FDA violated the APA. Id. Ipsen alleged two injuries-in-fact: (1) “that the regulation of Somatuline Depot as a drug rather than as a biological product exposes it to a greater risk of competition”; and (2) “that regulation under the FDCA entitles it to less information about potential patent infringement than would regulation under the PHSA, which hinders its ability to defend its manufacturing patents.” Id. at *4. The court found that both injuries were “too speculative to establish Article III standing.” Id. Thus, the court did not reach the merits of Ipsen’s argument regarding the framework for evaluating a “protein” under the BPCIA.
V. Antitrust and Competition
In 2021, we continued to monitor antitrust and competition-related litigation and developments concerning biologics, most notably AbbVie’s Humira® (adalimumab) and Johnson & Johnson’s Remicade® (infliximab). Both of these biologic products have been the targets of antitrust claims in the courts, and Humira® has also come under greater scrutiny by lawmakers.
Humira® (adalimumab) Developments
In March 2019, indirect purchasers of Humira® (adalimumab) brought a class action lawsuit against AbbVie alleging that AbbVie violated federal antitrust law by building a “thicket” of patents that prevented would-be challengers from entering the market with lower-cost biosimilar alternatives and entering into anticompetitive “pay-for-delay” and geographic market allocation agreements with adalimumab biosimilar manufacturers. (1:19-cv-01873 N.D. Ill.) In June 2020, the district court dismissed the complaint based on findings that AbbVie’s conduct was immune from antitrust liability under the Noerr-Pennington doctrine and that the plaintiffs failed to plausibly allege the existence of an agreement that restrained competition. In re Humira (Adalimumab) Antitrust Litig., 465 F. Supp. 3d 811 (N.D. Ill. 2020). In July 2020, the plaintiffs appealed the dismissal to the 7th Circuit. (No. 20-2402.) On February 25, 2021, a panel consisting of Circuit Judges Frank H. Easterbrook, Diane P. Wood, and Thomas L. Kirsch II heard oral arguments. A decision remained pending at the end of 2021.
On May 18, 2021, the U.S. House of Representatives Committee on Oversight and Reform held a hearing to investigate AbbVie’s pricing and business practices, including with respect to its blockbuster anti-inflammatory biologic, Humira® (adalimumab). AbbVie CEO Richard Gonzalez testified at the hearing, and the lawmakers inquired into a number of topics, such as AbbVie’s projections for Humira® biosimilar competition by 2017, AbbVie’s settlement agreements with adalimumab biosimilar developers, biosimilar competition in Europe as compared to the United States, and rebate programs and formulary placement agreements. On the same day, Chairwoman Carolyn Maloney also released a staff report describing AbbVie’s alleged efforts to delay competition and raise prices for Humira® and sent a letter to FTC calling for a “formal inquiry into whether AbbVie’s anticompetitive practices violated the law.” Another report was released by the Committee on Oversight and Reform on December 10, 2021, calling out AbbVie for engaging in numerous anticompetitive activities with respect to Humira®: “product hopping and evergreening,” “shadow pricing,” filing at least 257 patents for Humira®, and more.
Finally, as noted above, adalimumab biosimilar manufacturer Alvotech has filed affirmative defenses and counterclaims in BPCIA litigation alleging, inter alia, unclean hands and patent misuse tied to AbbVie’s alleged anticompetitive conduct. (See, e.g., 1:21-cv-02258 N.D. Ill. at Dkt. 60.) These allegations have not yet been substantively addressed by the court.
Remicade® (infliximab) Developments
Over the past several years, multiple suits have been filed alleging that Janssen and Johnson & Johnson engaged in a scheme to protect Remicade® (infliximab) in violation of federal antitrust law and maintained market share and pricing for Remicade® through anticompetitive devices, including exclusionary contracts, bundling, and coercive rebates.
The first suit was brought in 2017 by Pfizer, maker of the Remicade® biosimilar Inflectra®. (17-cv-04180 E.D. Pa.) After numerous discovery delays caused by the ongoing COVID-19 pandemic, on July 20, 2021, the parties filed a stipulation of dismissal. (Dkt. 167.) Various news outlets have reported that the parties settled their dispute and Pfizer has confirmed that it will continue to sell Inflectra® in the United States.7
A class action lawsuit by indirect purchasers (17-cv-04326 E.D. Pa.) and a suit brought by Walgreen and Kroger (18-cv-02357 E.D. Pa.) remain pending. These cases, also delayed by COVID-19, are proceeding through fact discovery.
VI. Post-Grant Challenges at the PTAB
Biologic and biosimilar activity at the Patent Trial and Appeal Board (PTAB) increased in 2021 compared to 2020. Only two biologic inter partes review (IPR) petitions and one post grant review (PGR) petition were filed in 2020. In 2021, this rose to eighteen IPR and five PGR biologic petitions, some of which are noted below.8
- Neulasta® (pegfilgrastim): In February 2021, Hospira and Pfizer filed an IPR against Amgen concerning U.S. Patent No. 8,273,707, directed to methods of protein purification requiring a certain combination of salts at certain concentrations, which Amgen asserted in pending district court litigation discussed above (20-cv-00201 D. Del.). (IPR2021-00528.) The PTAB granted institution in August 2021.
- Eylea®/Zaltrap® (aflibercept): In 2021, several parties filed IPR and PGR petitions against Regeneron concerning patents generally directed to vascular epithelial growth factor (“VEGF”) antagonist formulations suitable for intravitreal administration and methods of using VEGF antagonists to treat angiogenic eye disorders.
- In January 2021, Chengdu Kanghong Biotechnology Co., Ltd. brought an IPR and a PGR against a formulation patent (U.S. Patent No. 10,464,992) and a method of treatment patent (U.S. Patent No. 10,828,345), respectively. (IPR2021-00402; PGR2021-00035.) Neither challenge reached an institution decision, as Chengdu terminated both proceedings in June 2021.
- In May 2021, Mylan filed two IPRs against two method of treatment patents, U.S. Patent Nos. 9,669,069 and 9,254,338. (IPR2021-00880, -00881.) The PTAB granted institution of both IPRs in November 2021. Subsequently, in December 2021, Celltrion and Apotex filed petitions substantially similar to Mylan’s petitions and moved for joinder with the instituted IPRs. (IPR2022-00257, -00258, -00298, -00301.)
- In September 2021, Celltrion filed a PGR against a patent claiming formulations of VEGF antagonist fusion proteins (U.S. Patent No. 10,857,231). (PGR2021-00117.) The PTAB has not yet reached an institution decision.
- Actemra® (tocilizumab): Over the course of 2021, Fresenius Kabi filed six IPRs against Chugai Seiyaku Kabushiki Kaisha concerning five patents generally directed to methods for treating interleukin-6 (IL-6) related diseases. (IPR2021-01024, -01025, -01288, -01336, -01542; IPR2022-00201.) The PTAB has instituted IPR2021-01024 (challenging claims in U.S. Patent No. 7,521,052 to methods of treatment) and IPR2021-01025 (challenging claims in U.S. Patent No. 10,744,201 to methods of treatment). The PTAB has not yet reached institution decisions in the remaining four proceedings.
- Abecma® (idecabtagene vicleucel): In September 2021, Janssen filed an IPR against the United States as represented by the Secretary of Health and Human Services concerning a patent generally directed to chimeric antigen receptors (CARs) directed against B-cell Maturation Antigen (BCMA) (U.S. Patent No. 9,765,342). (IPR2021-01484.) The PTAB has not yet reached an institution decision.
Several disputes also reached resolution this year. For example:
- Humira® (adalimumab): In January 2021, the PTAB denied Fresenius Kabi’s request for rehearing of its decision denying institution of PGR of Coherus’s U.S. Patent No. 10,155,039—which is directed to stable aqueous formulations of adalimumab. (PGR2019-00064.) Both Fresenius Kabi and Coherus have proposed Humira® biosimilars and have settled with AbbVie with launch dates in 2023.
- Enhertu® (trastuzumab deruxtecan): In June 2021, the PTAB denied institution in two PGRs that Daiichi Sankyo, Inc. and AstraZeneca Pharmaceuticals, LP brought against SeaGen, Inc. concerning a patent related to antibody-drug conjugates (U.S. Patent No. 10,808,039). (PGR2021-00030, -00042.)
- Neupogen® (filgrastim) and Neulasta® (pegfilgrastim):
- In July 2021, the PTAB denied institution in an IPR that Lupin brought against Amgen concerning U.S. Patent No. 9,856,287, which is related to a method of refolding proteins expressed in non-mammalian cells. (IPR2021-00326.) Amgen previously asserted this patent in BPCIA litigation related to filgrastim and pegfilgrastim biosimilars against Apotex (0:18-cv-61828 S.D. Fla.), Kashiv (Adello) (2:18-cv-03347 D.N.J.), and Tanvex (3:19-cv-01374 S.D. Cal.).
- In November 2021, the PTAB denied Amgen’s request for director review and vacatur of the PTAB’s final written decision (as amended) in an IPR filed by Apotex finding claims 1–24 of U.S. Patent No. 8,952,138 (the “’138 patent”), titled “Refolding Proteins Using a Chemically Controlled Redox State,” unpatentable. (IPR2016-01542.) This request followed the Supreme Court’s decision in United States v. Arthrex, Inc., 141 S. Ct. 1970 (2021), and the Federal Circuit’s subsequent order giving Amgen the opportunity to request Director rehearing of the final written decision. Amgen had asserted the ’138 patent in BPCIA litigation, including against Kashiv (Adello) related to a Neupogen® biosimilar (2:18-cv-03347 D.N.J.) and against Apotex related to a Neulasta® biosimilar (0:15-cv-61631 S.D. Fla.).
In addition, the Federal Circuit decided several appeals from PTAB decisions in 2021, for example:
Ajovy® (fremanezumab-vfrm): In August 2021, the Federal Circuit issued two precedential decisions and one non-precedential decision related to biologic patents owned by Teva. The Court affirmed the PTAB’s decisions invalidating certain claims relating to antibodies and upheld other claims relating to methods of using those antibodies. See Eli Lilly & Co. v. Teva Pharms. Int’l GmbH, 8 F.4th 1331 (Fed. Cir. 2021) (affirming final written decisions in IPR2018-01710, -01711, and -01712 finding that petitioner had failed to demonstrate the challenged claims were unpatentable); Teva Pharms. Int’l GmbH v. Eli Lilly & Co., 8 F.4th 1349 (Fed. Cir. 2021) (affirming final written decisions in IPR2018-01422, -01423, and -01425 finding claims unpatentable due to obviousness); Teva Pharms. Int’l GmbH v. Eli Lilly & Co., 856 F. App’x 312 (Fed. Cir. 2021) (affirming final written decisions in IPR2018-01424, -01426, and -01427 finding claims unpatentable due to obviousness). Teva had asserted these patents in the District of Massachusetts against Eli Lilly in litigation, still ongoing, related to Teva’s migraine treatment Ajovy® (fremanezumab-vfrm) and Eli Lilly’s migraine treatment Emgality® (galcanezumab-gnlm). (See 1:18-cv-12029 D. Mass. at, e.g., Dkt. 1, 160.)
Lantus® (insulin glargine): In December 2021, the Federal Circuit issued three non-precedential decisions affirming the PTAB’s decisions finding unpatentable claims relating to injection devices for administering medicinal products. See Sanofi-Aventis Deutschland GmbH v. Mylan Pharms., Inc., No. 2020-1871, 2021 WL 6137374 (Fed. Cir. Dec. 29, 2021) (per curiam) (addressing IPR2018-01670, -01675, -01676, and -01678, and IPR2019-00122 and-00979); Sanofi-Aventis Deutschland GmbH v. Mylan Pharms., Inc., No. 2020-2066, 2021 WL 6137375 (Fed. Cir. Dec. 29, 2021) (per curiam) (addressing IPR2018-01679, -01680, and -01682); Sanofi-Aventis Deutschland GmbH v. Mylan Pharms., Inc., No. 2020-2071, 2021 WL 6138219 (Fed. Cir. Dec. 29, 2021) (per curiam) (addressing IPR2018-01684). Viatris issued a press release the same day, stating “it is pleased with decisions issued today which affirm the U.S. Patent and Trademark Appeal Board’s prior rulings that found the challenged claims of Sanofi’s Lantus® SoloSTAR® device patents, U.S. Patent Nos. 9,603,044, 8,992,486, 9,526,844, 9,604,008, and 8,679,069, unpatentable.”
Finally, also of note, in August 2021, USPTO published an updated report on AIA proceedings involving Orange Book-listed patents and biologic patents. According to the report, through June 2021, 2% of all AIA petitions challenged biologic patents, and the institution rate for biologic patents was 55%. The report states that challenges to both Orange Book and biologic patents have decreased over time.
VII. Conclusion
While many traditional metrics of biosimilar activity (FDA approvals, biosimilar launches, new case filings) remained low in 2021, many stakeholders (including regulatory agencies and legislators) remain focused on the promise of biosimilars to increase access and reduce pharmaceutical costs. Increasing biosimilar market penetration shows that this can be achieved.
We have many new biosimilar developments to look forward to in 2022 and beyond. If COVID-19 restrictions and delays are mitigated or eliminated, we may see additional biosimilar clinical trials, new biosimilar BLAs, and increasing FDA approvals. With the recent launch of the first interchangeable, Semglee®, we will start seeing how interchangeables function in the market. With the pending antitrust appeal at the Seventh Circuit, the ongoing Congressional focus on Humira®, and Alvotech’s allegations of unclean hands and patent misuse, we will also likely get some guidance on the line between fair competition and unlawful anticompetitive behavior in the biosimilars space.
[1] See, e.g., Amgen Biosimilars’s 2021 Biosimilar Trends Report (“Amgen Biosimilars Report”), USA-CBU-80962_Amgen-2021-Biosimilar-Trends-Report.pdf (amgenbiosimilars.com) (last accessed Jan. 9, 2022); FDA Biosimilars Approvals, Biosimilars Council (Sept. 20, 2021) https://biosimilarscouncil.org/resource/fda-biosimilars-approvals/ (last accessed Jan. 9, 2022).
[2] Mylan first filed for approval of Semglee® as an NDA. As of March 23, 2020, Mylan’s NDA for Semglee® was “deemed” to be a BLA under § 351(a) (i.e., not a biosimilar application under § 351(k)). Semglee® was launched in August 2020 under that § 351(a) BLA, and Mylan pursued interchangeable status through a separate BLA, which was ultimately approved in July 2021.
[3] Approval time is calculated from the first BLA submission date, not any resubmission date.
[4] Mylan filed its New Drug Application (NDA) for Semglee® on April 27, 2017, and on March 23, 2020, Mylan’s NDA was deemed to be a BLA.
[5] Amgen Biosimilars Report.
[6] This case was originally filed in the Eastern District of Virginia with case number 1:21-cv-00589. On May 12, 2021, the case was transferred intradistrict from the Alexandria Division to the Norfolk Division of the Eastern District of Virginia (2:21-cv-00265). The case was then transferred to the Northern District of Illinois on October 22, 2021 and assigned case number 1:21-cv-05645 N.D. Ill.
[7] See, e.g., https://www.fiercepharma.com/pharma/pfizer-johnson-johnson-agree-to-a-settlement-terms-undisclosed-biosimilar-lawsuit (last accessed Jan. 9, 2022).
[8] A USPTO report, issued in August 2021, reports slightly different numbers for total biologic IPRs and PGRs per year, likely as a result of categorizing certain types of patents differently. https://www.uspto.gov/sites/default/files/documents/PTABOBbiologicpatentstudy8.10.2021draftupdatedthruJune2021.pdf (last accessed Jan. 9, 2022). However, the general trends are the same.
The opinions expressed are those of the authors on the date noted above and do not necessarily reflect the views of Fish & Richardson P.C., any other of its lawyers, its clients, or any of its or their respective affiliates. This post is for general information purposes only and is not intended to be and should not be taken as legal advice. No attorney-client relationship is formed.